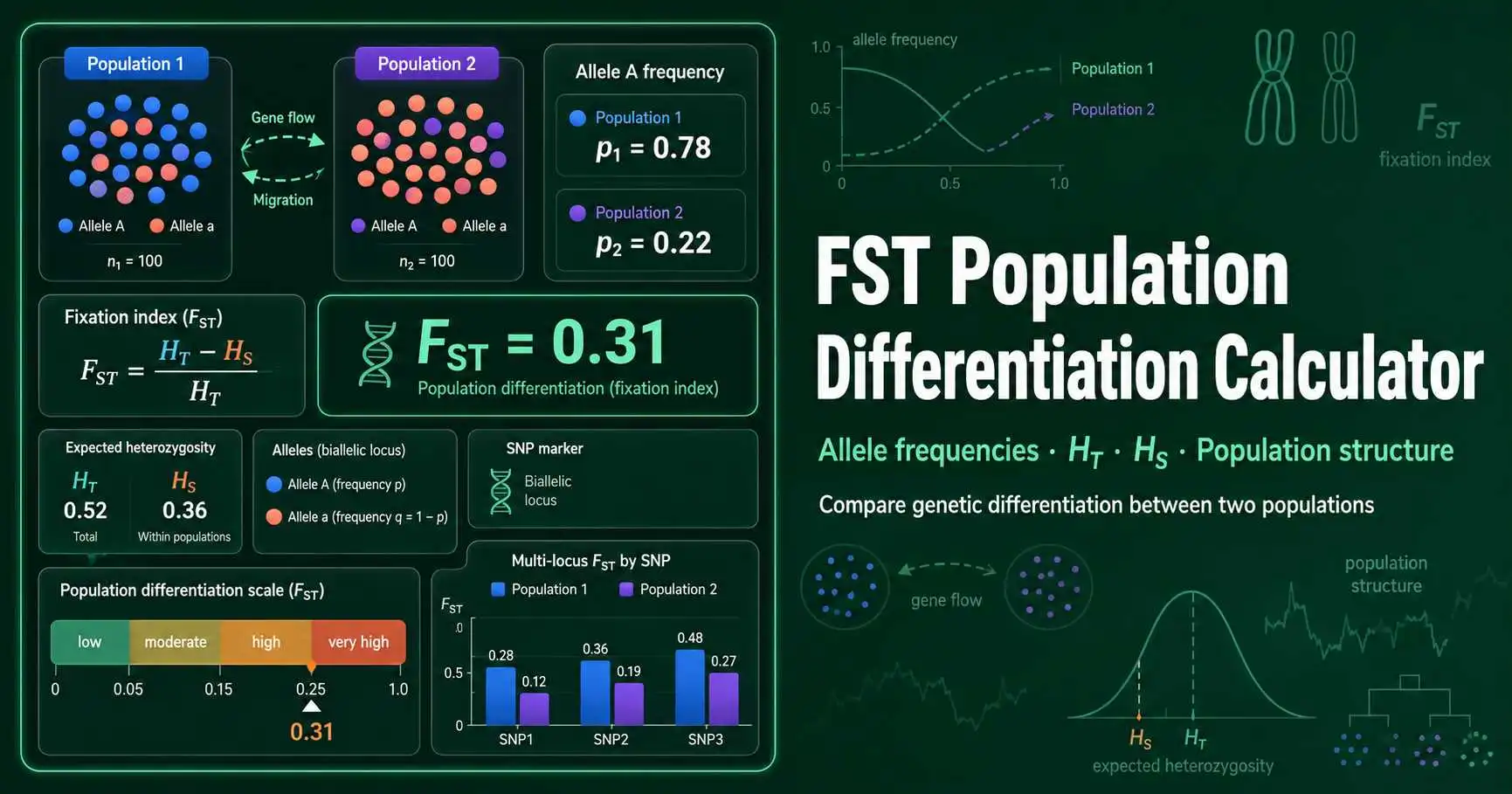
FST is a fixation index that describes genetic differentiation among populations. Sewall Wright developed the F-statistics framework to measure how population subdivision changes genotype and allele-frequency patterns. In practical terms, FST asks how much total genetic diversity comes from allele-frequency differences between populations.
A value near 0 means the sampled populations carry similar allele frequencies. A value near 1 means the populations show strong separation at the markers tested. Holsinger and Weir describe FST as the proportion of genetic diversity due to allele-frequency differences among populations. Read their population-structure review.
This calculator uses a heterozygosity-based biallelic model. It works well for teaching SNP-like markers because every locus uses allele A frequency in population 1 and population 2. Researchers who analyze large sequencing panels often use software that adds confidence intervals, missing-data handling, and Weir-Cockerham estimators.