Oligo Secondary Structure: Hairpins and Dimers
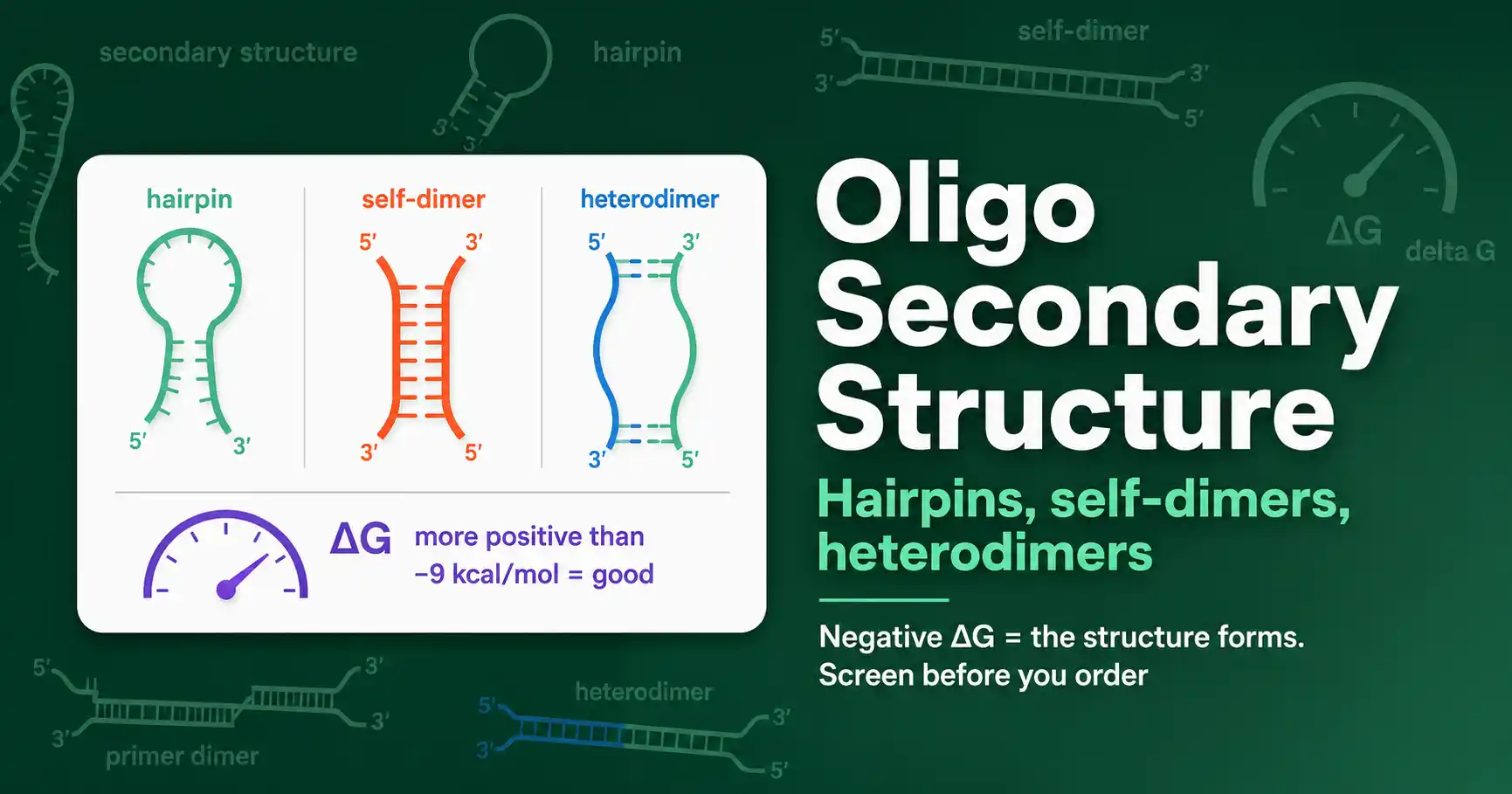
Oligo secondary structures are unwanted shapes an oligo forms by pairing with itself or another oligo instead of binding its target. The three that matter are hairpins, self-dimers, and heterodimers. Each ties up primer that should be working, so they cause weak bands, primer-dimer artifacts, and failed reactions. You catch them by checking the binding energy, the ΔG, of every possible structure before you order.
This guide explains each structure, how to read the ΔG value that predicts whether it forms, and how to fix an oligo that fails the check. It is the analyzer-facing capstone of this series, the detailed version of the screening step in our guide on PCR primer design guidelines.
What Secondary Structure Means
Secondary structure is any folded or paired shape an oligo takes other than a straight single strand. It happens because an oligo can base-pair with sequences that are complementary to itself, and those self-pairings compete with the pairing you actually want.
The problem is competition for the primer. An oligo locked in a hairpin or stuck to another oligo is not available to bind the template, so secondary structure reduces the effective primer concentration. Worse, some structures get amplified themselves, producing artifacts that consume reagents and obscure the real product. So secondary structure is not a cosmetic concern; it directly lowers yield and specificity.
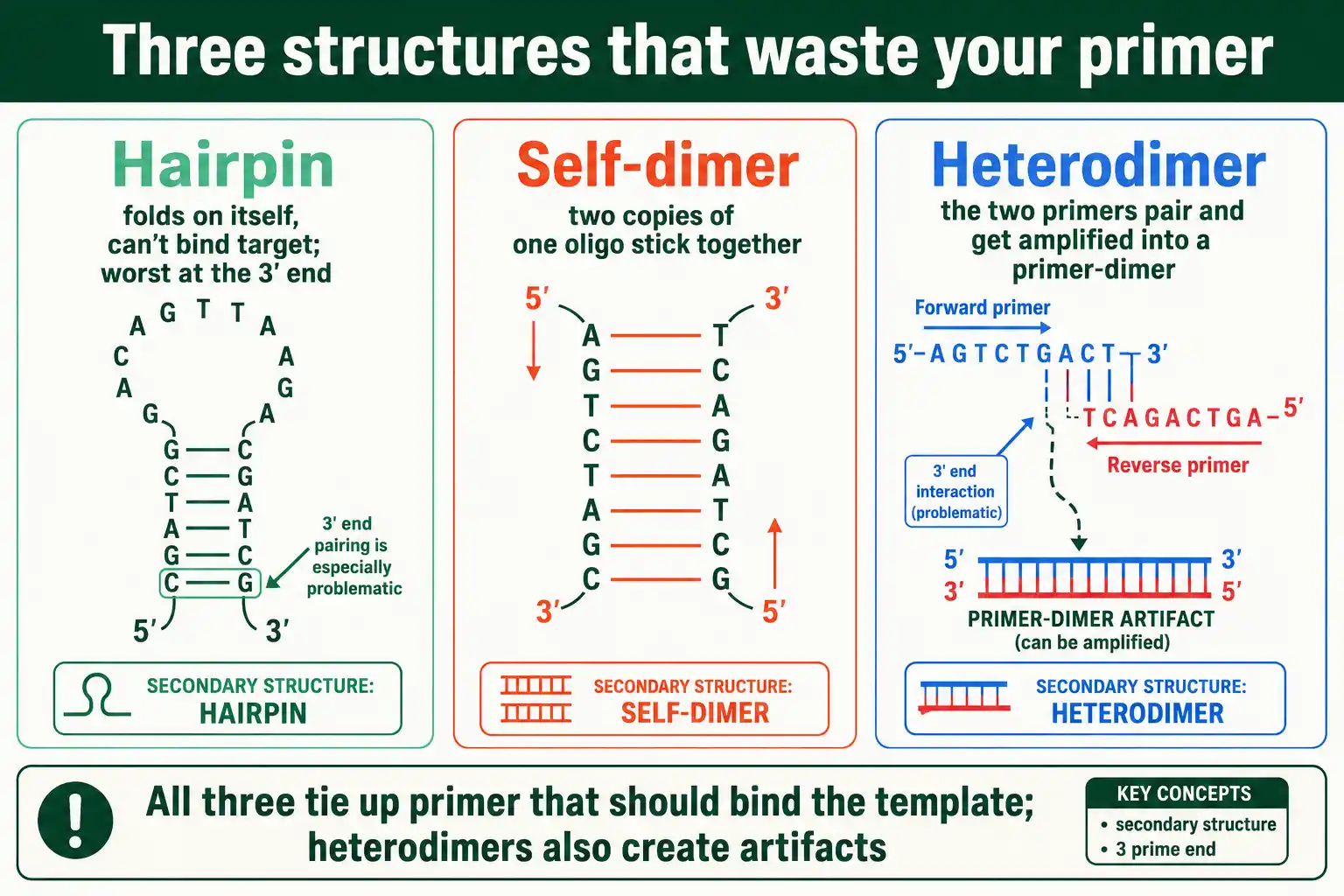
Three structures cause nearly all the trouble: hairpins, where one oligo folds on itself; self-dimers, where two copies of the same oligo pair; and heterodimers, where two different oligos, usually the forward and reverse primers, pair with each other. The rest of this guide takes them in turn.
Hairpins
A hairpin forms when an oligo folds back on itself, pairing an internal stretch with a complementary stretch elsewhere in the same strand. The result looks like a loop with a paired stem.
Hairpins matter because a folded oligo cannot bind its target. The oligo is sequestered in its own loop, so it is unavailable until the hairpin melts, which lowers the effective primer concentration and can stop the reaction from priming. A hairpin at the 3' end is the most damaging, because it blocks the exact end the polymerase needs to extend from. Hairpins arise from internal self-complementarity, so an oligo with a stretch of sequence that matches its own reverse complement a few bases away is prone to them. Designing the sequence to break up such internal complementarity is the fix, which usually means shifting the primer a few bases along the template.
Self-Dimers and Heterodimers
Dimers form when two oligo molecules pair with each other instead of the template. A self-dimer is two copies of the same oligo pairing; a heterodimer, also called a cross-dimer, is the forward and reverse primers pairing with each other.
Both waste primer the same way hairpins do, but heterodimers carry an extra danger. When the two primers pair through their 3' ends, the polymerase extends each one using the other as a template, amplifying the dimer itself. This primer-dimer product competes with the real target, often winning because it is short and amplifies fast, and it shows up as a bright low-molecular-weight band that drains the reaction. This is why 3' complementarity between primers is the single worst feature a primer pair can have. The fix is to avoid any complementarity between the primers' 3' ends, redesigning one primer if needed.
Reading the ΔG Value
The key number for any secondary structure is its ΔG, the Gibbs free energy of formation, which predicts whether the structure will actually form. Understanding the sign and scale of ΔG is the heart of using an analyzer.
ΔG measures the energy released when a structure forms, so a more negative ΔG means a more stable, more favorable, more likely structure. A ΔG near zero or positive means the structure is unstable and essentially will not form. This is the rule to internalize: negative ΔG is bad, because it means the unwanted structure forms readily, while positive ΔG is good, because the structure does not form at all. The value comes from the same thermodynamics that set melting temperature, ΔG = ΔH − TΔS, applied to the folded or paired conformation.
This shared thermodynamic basis links secondary structure to the properties covered in our guide on GC content and melting temperature. A GC-rich oligo, with its stronger three-hydrogen-bond pairs, not only has a higher melting temperature but also forms more stable secondary structures, because the same base-pairing that stabilizes the duplex stabilizes a hairpin or dimer. So a high-GC primer that looks attractive for its tight target binding can be more prone to folding on itself, which is one reason GC extremes are discouraged in primer design. The ΔG of a structure rises and falls with temperature too, which is why a structure stable at a low annealing temperature may melt away at a higher one.
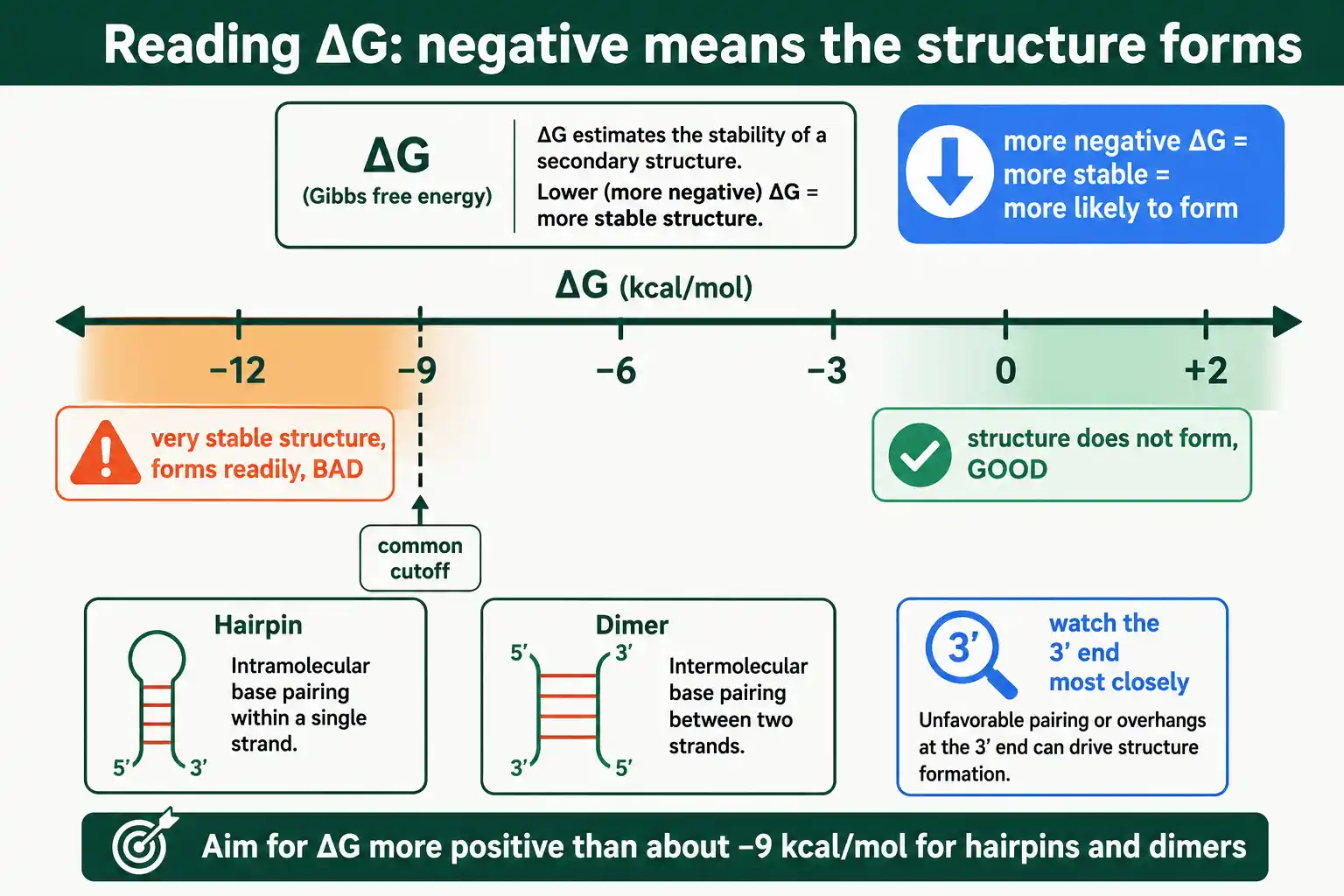
A widely used threshold makes this practical. For overall hairpins and dimers, aim for a ΔG more positive than about −9 kcal/mol; structures more negative than that form readily enough to cause problems. The MIT synthetic biology primer design guidance uses exactly this −9 kcal/mol cutoff for self-dimers, hairpins, and heterodimers. Finer guidance tightens the limit for the 3' end, where structures do the most harm, and for sensitive applications like qPCR.
ΔG Thresholds at a Glance
Different structures and positions tolerate different ΔG values. The table collects commonly used cutoffs.
| Structure | Position | Tolerated ΔG (more positive than) |
|---|---|---|
| Hairpin | 3' end | about −2 kcal/mol |
| Hairpin | Internal | about −3 kcal/mol |
| Self-dimer | 3' end | about −5 kcal/mol |
| Self-dimer | Internal | about −6 kcal/mol |
| Heterodimer | 3' end | about −5 kcal/mol |
| Any structure | Overall | about −9 kcal/mol |
The pattern in the table is consistent: 3' structures are tolerated only at weaker (more positive) ΔG than internal ones, because the 3' end is where priming happens. For routine PCR these thresholds can be relaxed slightly; for qPCR, where small artifacts distort quantification, hold to the stricter end, around −3 kcal/mol for hairpins and −6 for dimers. The ResearchGate discussion of permissible ΔG values collects practitioner consensus on these numbers.
Beyond Primers: Probes and Templates
Secondary structure matters for more than PCR primers. Any oligo that must bind a target competes with its own folding, so the same checks apply to probes, and template structure can interfere too.
Hybridization probes, including the dual-labeled probes used in quantitative PCR, must avoid hairpins for the same reason primers must: a folded probe cannot bind its target, and for some probe chemistries an unintended fold can also bring a fluorophore and quencher together or apart in ways that ruin the signal. Probes are screened against the same ΔG thresholds, often more strictly, because a quantitative readout is sensitive to small inefficiencies. The IDT guidance on checking oligos for hairpins and dimers applies the same workflow to probes and primers alike.
Template secondary structure is the other side of the problem. A target region that itself folds into a stable hairpin can block a primer or probe from binding, even when the oligo is well designed. This is common in GC-rich templates and is one reason some regions are hard to amplify. When a well-designed primer still fails, suspect template structure, and consider additives like DMSO that relax it, or move the primer to a less structured region. The oligo is only half of the binding event; the target has to cooperate too.
How to Check an Oligo
You check for secondary structure by computing the ΔG of every possible hairpin and dimer, which is a job for a tool, not hand calculation. The check takes seconds and saves failed reactions.
The workflow is straightforward. Enter the oligo sequence, and for dimers enter the second primer too, then read the reported structures and their ΔG values. Our oligo analyzer reports the predicted hairpins, self-dimers, and heterodimers along with each one's ΔG, so you can see at a glance whether any structure crosses the threshold. Compare each reported ΔG against the cutoffs above, paying closest attention to any structure involving the 3' end. If everything is more positive than the relevant threshold, the oligo passes; if not, it needs fixing.
Specifying the right reaction conditions improves the prediction. Because ΔG depends on temperature and salt, entering the actual annealing temperature, sodium concentration, and magnesium concentration gives a result that reflects your real reaction rather than default conditions. This matters most for borderline cases near a threshold.
How to Fix a Problem Oligo
When an oligo fails the secondary-structure check, several fixes can rescue it. The right one depends on which structure is the problem.
The most general fix is to shift the primer. Moving the primer a few bases along the template changes its sequence and usually breaks up the self-complementarity causing a hairpin or dimer, while still amplifying the same region. For a heterodimer, redesign whichever primer can move without compromising the amplicon. A second option is to add a few bases to the 3' end: if a self-dimer pairs through the 3' end, extending the end with a couple of non-complementary bases stops the polymerase from extending the dimer, even though the pairing remains. A third option is a 5' tail or flap, adding non-complementary A/T-rich sequence at the 5' end, which can improve performance for a sequence whose own composition makes it structure-prone.
Adjusting conditions helps too. Running the reaction at a higher annealing temperature melts weak secondary structures that would form at lower temperatures, so a borderline oligo sometimes works simply by raising the temperature. But the durable fix is in the sequence: an oligo designed without strong self-complementarity, and especially without 3' complementarity to its partner, avoids the problem from the start. Screening before ordering is always cheaper than troubleshooting after.
Frequently Asked Questions
What does a negative ΔG mean for an oligo?
A negative ΔG means a secondary structure is stable and likely to form, which is bad. The more negative the ΔG, the more readily the hairpin or dimer forms and the more it interferes with the reaction. A ΔG near zero or positive means the structure will not form, which is what you want. As a rule, keep hairpin and dimer ΔG more positive than about −9 kcal/mol.
Why are 3' end secondary structures the worst?
Because the 3' end is where DNA polymerase begins extension. A hairpin or dimer involving the 3' end either blocks priming, if it sequesters the end, or gets actively amplified, if two primers pair through their 3' ends and extend off each other. The latter creates primer-dimer artifacts that compete with the real product, which is why 3' complementarity is the most damaging feature in a primer pair.
How do I get rid of a primer dimer?
Identify the complementary region with an analyzer, then redesign to remove it, usually by shifting the primer a few bases so its 3' end is no longer complementary to itself or its partner. Adding non-complementary bases to the 3' end or raising the annealing temperature can help, but the reliable fix is eliminating the 3' complementarity in the sequence before ordering.
Catching Problems Before They Cost You
Oligo secondary structures, hairpins, self-dimers, and heterodimers, all do the same damage in different ways: they tie up primer that should be binding the target, and heterodimers can be amplified into artifacts that crowd out the real product. The 3' end is the danger zone for all of them, because that is where the polymerase works. The single number that predicts trouble is ΔG, where more negative means a more stable, more likely, more problematic structure.
Screening is fast and worth doing on every oligo: compute the ΔG of each possible structure, compare it against the thresholds, and redesign anything that crosses them, especially at the 3' end. With this final check, the toolkit is complete, from understanding what an oligo is, through quantifying and diluting it, to designing one that binds cleanly and folds the way you want. To revisit how these properties are computed for any sequence, our explainer on what an oligonucleotide is returns to the foundations behind the whole workflow.